
| 의료진 | 정지혁 |
| 관련 질환명 | Noack증후군(Noack syndrome ) 첨두합지증 5형( Acrocephalosyndactyly type 5 ) 두개안면-골격-피부 이형성증( Craniofacial-skeletal-dermatologic dysplasia ) |
| 증상 | 두개골조기유합에 의한 단두(短頭), 위턱뼈발육부전에 의한 깊이가 얕은 눈주위뼈와 안구돌출, 그리고 넓은 엄지손가락과 엄지발가락 등의 세 가지 특징을 나타냄 |
| 관련 클리닉 |
파이퍼 증후군 (Pfeiffer syndrome : ICD-10 code : Q87.0|)이란?
파이퍼증후군은 두개골조기유합에 의한 단두(短頭), 위턱뼈발육부전에 의한 깊이가 얕은 눈주위뼈와 안구돌출, 그리고 넓은 엄지손가락과 엄지발가락 등의 세 가지 특징을 나타내며, 상염색체우성으로 유전됩니다. 지능은 대개 정상인 것으로 알려져 있습니다. 파이퍼 증후군은 두개골 조기 유합증의 하나로, 1964년 독일의 유전학자인 Rudolf Arthur Pfeiffer에 의해 ‘우성 유전 첨두합지증(dominant hereditary acrocephalosyndactyly)’이라고 기술되었습니다. 당시 Pfeiffer는 이 증후군을 두개골 봉합선의 조기 융합으로 인한 비정상적인 두개골 모양, 넓은 엄지손가락과 큰 엄지발가락, 손과 발의 다양한 정도의 합지증을 보이는 질환으로 정의하였습니다. 현재는 다양한 정도의 관상 봉합(coronal suture)의 조기 유합과 안면 구조의 이상으로 뾰족하고 납작한 머리, 상악골의 저형성, 턱의 돌출, 안구 돌출, 양안 격리, 사시, 낮은 콧잔등, 후비공 폐쇄증(choanal atresia), 기관지 연골의 기형, 요골-상완골의 유합, 손, 발의 기형, 수두증을 보이는 질환으로 정의됩니다.
두개골 조기 유합증의 발생율은 2,000-2,500명당 1명이며 이 중 파이퍼증후군의 발생 빈도는 100,000명당 1명으로 드문 질환입니다.
파이퍼증후군의 증상은 어떤 것들이 있나요?
발병 원인과 그 기전은 밝혀져 있지 않으며, 일부 연구자들은 다양한 외상의 결과로 뼈로의 혈액공급이 과다하게 증가하거나 조골세포와 파골세포 사이의 균형이 깨지는 것이 원인일 것이라고 추측하고 있다. 파골세포가 조골세포보다 우세하면 뼈 상실은 뼈 생산보다 앞서게 되어 뼈 용해가 일어날 수 있다.
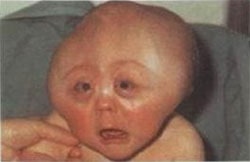
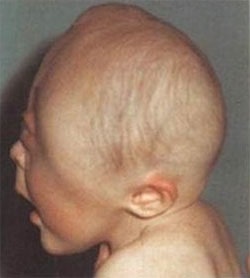
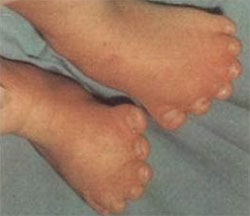
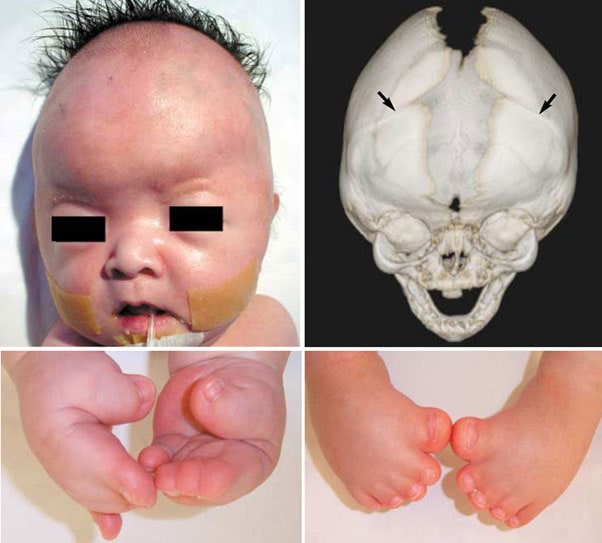
파이퍼 증후군 환자는 두개골 조기 유합(Craniosynostosis)으로 인해 비정상적인 두개골 모양과 안면 중앙부의 저형성으로 인한 기형을 나타냅니다. 주로 관상 봉합선(전두골과 두정골을 나누는 봉합선)이 조기에 유합되어 두개골이 비정상적으로 자랍니다.
신체적인 특징은 Crouzon증후군 등의 두개골 조기 유합증과 유사하면서 넓은 엄지손가락과 발가락이 나타납니다. 특징적인 안면 모양으로 튀어나온 이마, 함몰된 광대뼈, 작은 상악골, 낮은 콧잔등, 튀어나온 턱을 보이며 안구가 현저히 돌출되어 있으면서 양안의 간격이 넓습니다. 작은 코나 높은 궁상의 구개, 또는 교합 부전으로 인해 공명이 있는 언어 장애가 생길 수 있고, 구개열도 있을 수 있습니다. 이 환자들은 안근의 불균형으로 시각적 장애를 가질 수 있으며, 중이염이 반복되어 청력의 상실이 있을 수도 있다. 넓은 엄지손가락과 큰 발가락을 보입니다.
증상에 따라 1형, 2형, 3형의 3가지 아형이 있습니다. 파이퍼증후군 1형이 가장 경미하고 3형은 가장 심한 임상 양상을 보입니다. 1형은 Pfeiffer에 의해 기술된 전형적인 파이퍼증후군으로 예후가 좋고 지능이 정상이며 상염색체 우성으로 유전됩니다. 2형에서는 팔꿈치 관절이 강직되어 있으며, 관상 봉합선과 시상 봉합선(뇌의 좌우를 분리해주는 봉합선)이 모두 일찍 닫혀 clover-leaf 모양의 두개골을 보이며 심한 안구 돌출과 손과 발의 기형이 동반되며 예후가 나쁩니다. 3형은 2형과 비슷한 양상으로 보이나 cloverleaf 모양의 두개골은 보이지 않습니다.
파이퍼증후군의 심각한 합병증은 호흡기계 문제와 수두증입니다. 호흡 문제는 기관지의 기형과 연관되어 있으며 일부 환자에서는 기관 절개를 필요로 합니다. 수두증은 치료하지 않으면 정신 발달 지연이 동반될 수 있습니다. 2형과 3형 파이퍼증후군 환자에서 심각한 합병증이 더 자주 발생하여 영아기를 넘기지 못하는 경우도 있으며 수두증이 동반되지 않더라도 발달 지연, 정신 지체, 경련이 흔하게 나타납니다. 1형 파이퍼증후군 환자의 지능은 보통 정상 범위에 속하나 2형과 3형에서는 약간의 정신 지체도 있을 수 있습니다.
치료는 어떻게 하나요?
파이퍼 증후군 환자는 성형외과, 신경외과, 정형외과, 이비인후과, 치과 등 각 분야의 전문의에 의한 정기적인 관찰과 치료가 필요합니다. 성형외과와 신경외과적인 수술이 여러 단계로 시행되는 것이 일반적이며 수술 시기와 방법은 환자 개인에 따라 다르다. 영유아기 때에는 뇌의 정상적인 성장을 위해 필요한 적당한 두개 내 공간을 갖도록 두개골의 골유합 봉합을 제거해 줍니다. 이 환자들은 성장기에 따라 적절한 두개골의 성형이 필요할 수도 있으며, 필요한 경우 안면 성형 수술도 시행합니다. 수술 후 완전히 정상의 모습을 찾기는 어려우나 상당히 호전될 수 있으며 수술의 시기가 중요합니다.
손가락과 발가락은 현저한 기형이 있음에도 불구하고 그 기능은 정상일 수 있으므로 성형 수술이 반드시 필요하지는 않다. 수두증, 기도 폐쇄, 청력 손실, 척추 기형은 빠른 시간 내에 치료가 필요합니다. 예후는 증상에 따라 다릅니다. 1형의 경우 경미한 증상을 보여 2형이나 3형보다 예후가 좋습니다.
참고문헌
Cunningham ML, Seto ML, Ratisoontorn C, Heike CL, and Hing AV. Syndromic craniosynostosis: from history to hydrogen bonds. Orthod Craniofac Res 2007;10:67-81.
본 콘텐츠의 저작권은 서울대학교병원에 있으며, 이를 무단 이용하는 경우 저작권법 등에 따라 법적 책임을 질 수 있습니다.
본 웹페이지에서 제공하는 내용은 참고사항일 뿐이며 게시물에 대한 법적 책임은 없음을 밝혀드립니다. 본인에 맞는 적절한 진단 및 치료를 위해서는 의사와 상담하시기 바랍니다.